
El CCAD ofrece el Seminario “Cómo correr AlphaFold2 en MendietaF2” Online, Libre y Gratuito Dictado por: Álvaro Schachner (FaMAF-UNC) y Juan Ignacio Folco (Ingeniería-UCC) Viernes 28/6, 10 a 11.30 hs https://meet.google.com/gsq-tcbc-msd
Categoría: Sin categoría
Jun 25 2024
Resultados del Reempadronamiento en CCAD
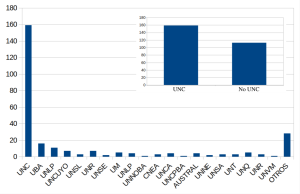
Inspirados por un procedimiento de reempadronamiento del NLHPC, en 2024 pedimos a todas/os las usuarias/os que indicaran que son usuarias/os activos. También preguntamos sobre sus trabajos científicos realizados, aportes al CCAD para su funcionamiento y qué proyectan para este año. Desde hace ya varios años que tenemos una activa y muy buena relación con nuestras …
May 28 2024
Circulando RAM a Bariloche

Hace poco recibimos partes de COSMA6, una supercomputadora del Reino Unido. Pasamos un décimo de estas memorias al Centro Átómico Bariloche para que puedan incrementar la cantidad de RAM de varios servidores de cómputo. Apenas recibimos la donación de Durham, vimos que teníamos más módulos de RAM de los que necesitabamos de manera inmediata. Para …
May 18 2024
Modelos de Lenguaje GRANDES en GPUs chicas
Como estudiantes de doctorado en Aprendizaje Automático en FaMAF-UNC, disponemos de capacidades de cómputo a través del CCAD. Aunque éstas están a la vanguardia a nivel nacional, aun así resultan insuficientes para entrenar modelos grandes de lenguaje. En este artículo técnico describimos experimentos para «pegar» dos NVIDIA A30 de 24 GiB y así poder entrenar …
Abr 22 2024
El CCAD renueva sus webs

En los últimos meses, estuvimos trabajando en la renovación de la wiki y la web, en conjunto con el rebranding a «UNC | Supercómputo». Problema El CCAD tenía un problema de identidad, tanto sus redes, como su web y su wiki no mantenían un estilo unificado, ni siquiera usaban el mismo tono violeta. Sumado a …
Abr 14 2024
La Fundación Vía Libre nos ayuda con Hardware

Todo suma y la Fundación Vía Libre nos da una mano con hardware variopinto que necesitamos. La diaria de hacer HPC no es comprar 15 chasis con 60 nodos, es otra cosa, un poco más precisa, más pequeña, pero fundamental para que todas las piezas encajen. ¿Qué hacemos con un riser PCIe, una NIC InfiniBand, …
Abr 05 2024
«Carpinchos» viaja a China para competencia internacional.
La semana del 8 de abril se desarrollará la onceava edición de la competenciaStudent Supercomputer Challenge de Asia Supercomputing en el marco de ASC24 AsiaSupercomputing. El equipo Carpinchos del Centro de Computación de Alto Desempeño (CCAD)de la UNC clasificó para la final quedando entre los 25 mejores de más de 300equipos de todo el mundo. …
Abr 05 2024
CCAD le da una segunda vida a COSMA6

La Universidad de Durham donó partes de COSMA6 al CCAD que serán usadas para mejorar las capacidades de red de alta velocidad y de memoria de nuestros clusters más viejos. Desde hace años que existe una relación muy fluida entre Durham University y la UNC a través de las y los investigadores del IATE-CONICET en …
Feb 07 2024
Todos los racks del CCAD con energía

La empresa PROIMA energizó 6 racks con 150 KVA, duplicando así la capacidad de alojar equipamiento de cómputo de alto desempeño. El costo de la obra aportados por el Rectorado de la UNC fue cercano a 40.000 dólares estadounidenses. En marzo de 2022 en una fructífera reunión con el entonces Rector de la UNC, el …
Feb 04 2024
Todo Serafín con 256 GiB de RAM

Supermicro, el proveedor de Serafín, terminó de realizar el cambio de memorias. Ahora los 60 nodos de cómputo tienen 256 GiB de RAM y el nodo cabecera 128 GiB, duplicando la memoria de la configuración original. En agosto de 2023 llegó un lote de memoria de recambio para reemplazar módulos DDR4-3200 de 8 GiB que …
HITOS


