
En marzo pusimos a disposición una partición y prioritaria para trabajos relacionados con COVID-19. Hubo tres proyectos que están haciendo uso de la infraestructura para simulaciones: dos de drug repurposing con diferentes enfoques, y uno sobre estrategias de cuarentena. Se utilizaron hasta ahora 54.000 horas/core de trabajo computacional.

Flyer realizado por Marcos Mazzini.
A partir del inicio del ASPO en Argentina, ofrecimos tiempo de cómputo prioritario para proyectos relacionados a COVID-19. Hubo tres proyectos que utilizaron los recursos computacionales del CCAD-UNC.
- Drug repurposing de la FCQ-UNC: screening virtual de para predecir la interacción de fármacos con proteínas y testear fármacos ya aprobados por la FDA con el objetivo de comprobar si alguno es efectivo contra el SARS-CoV-2.
- Drug repurposing del IIMT-Universidad Austral: el mismo objetivo que el anterior, pero con diferente base de datos de fármacos y la utilización de química cuántica para los cálculos de interacciones.
- Estrategias de cuarentena basados en circulación de personas de FaMAF-UNC.
Segundos usos a fármacos conocidos
El equipo de la FCQ-UNC formado por Marcelo Piuatti (Química Orgánica), Alfredo Quevedo (Farmacia), Rodrigo Quiroga (Química Teórica), Alexis Paz (Química Teórica), Sergio Ribone (Farmacia) y Marcos Villarreal (Química Teórica).
Los investigadores nos cuentan de qué se trata el proyecto.
El objetivo del proyecto es el reposicionamiento de fármacos (repurposing) para enfrentar al COVID-19. El repurposing consiste básicamente en encontrar un nuevo uso para un fármaco ya conocido. En nuestro caso estamos utilizando docking molecular y simulaciones de dinámica molecular para realizar las predicciones.
En una primera etapa hicimos una exploración de la capacidad de unión de los más de 3500 fármacos aprobados para uso humano por la FDA (Food and Drug Administration de EEUU) en las 26 proteínas que conforman el virus SARS-CoV-2. De un primer análisis se identificaron varias proteínas como potenciales blancos farmacológicos, como ser la proteína Spike, la Helicasa y la 2-O-MT.
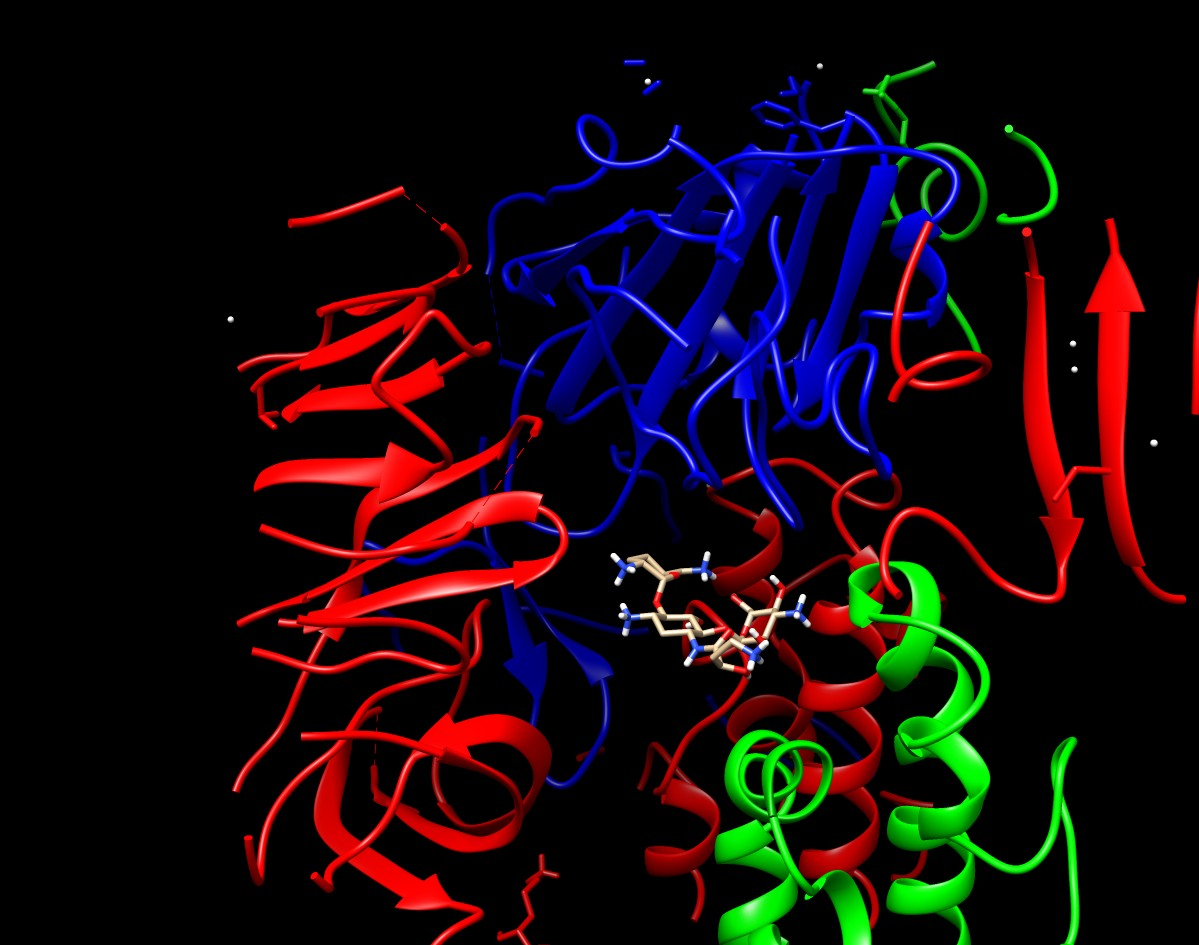
A mediados de Abril surgió una colaboración con el Dr. Cameron Abrams (Universidad de Drexel, Filadelfia) y el Dr. Andrés Finzi (Universidad de Montreal). Esta colaboración se centra en encontrar fármacos que mediante la inhibición de la proteína Spike impidan el ingreso del virus a la célula huésped. En la colaboración, el Dr. Abrams realiza simulaciones de dinámica molecular (MD) de la proteína Spike, nosotros realizamos docking y cálculos MM-GBSA sobre los resultados de la MD
(ver abajo el render estático y dinámico) y el Dr. Finzi ensaya experimentalmente las predicciones en un modelo de infección viral que ha puesto a punto en su laboratorio. Actualmente se han probado 7 compuestos con resultados un poco contradictorios. En un comienzo los ensayos mostraron potencial inhibitorio en varias de las drogas, mientras que en una segunda repetición fueron inactivos. En el laboratorio del Dr. Finzi se encuentran realizando nuevos ensayos al respecto.
Mientras aguardamos novedades estamos por retomar la idea original de estudiar las restantes proteínas del virus.
En la parte de docking, los investigadores hicieron un fork del proyecto iDock que denominaron Vinardo. Los investigadores explican que fué lo que cambiaron.
Nosotros le metimos mano, más del lado de la química que desde el lado de computación. Básicamente mejoramos la forma de estimar la energía de unión, que es el core del problema.
Vinardo no corría de manera eficiente en los 64 núcleos KNL de los nodos de Eulogia, por lo que el CPA Principal de CONICET Carlos Bederián transformó el paralelismo que hacía uso de boost::threads a OpenMP obteniendo una ganancia sustancial de desempeño.
Otra vuelta de tuerca al Drug Repurposing
El Laboratorio de “Diseño Computacional de Fármacos e Informática Biomédica” del Dr. Claudio Cavasotto (Instituto de Investigaciones en Medicina Traslacional, CONICET-Universidad Austral) publicó recientemente un trabajo usando la estrategia de reposicionamiento de fármacos, en el que se analizaron más de 11.000 compuestos, y se identificaron candidatos promisorios para COVID-19.
Juan Di Filippo, becario doctoral del Dr. Cavasotto estuvo utilizando MOPAC 2012 (Molecular Orbital PACkage), un paquete de química quántica (QC), para cálculos de proteínas del SARS-CoV-2 que genera el COVID-19. El trabajo fué además reseñado por la Universidad Austral, la Agencia Telam y el diario Clarín. Los autores nos comentan que,
La idea fue optar por una estrategia de reposicionamiento de fármacos, es decir encontrar un uso nuevo a un fármaco conocido. De esta forma, se ahorra mucho tiempo y se reducen los costos, ya que los fármacos conocidos ya han pasado las etapas de evaluación de toxicidad y propiedades farmacológicas. Ten en cuenta que el tiempo y costo total para desarrollar una nueva droga desde sus inicios se estima en +10 años y +2.5 billones de dólares.
Es importante marcar la diferencia con el trabajo realizado por el equipo de la FCQ-UNC. Una de las diferencias es la base de datos de moléculas, que fue construida por el Dr. Cavasotto a partir de fármacos aprobados para su uso o que están avanzadas en las etapas de evaluación.
Otra diferencia es que además de Spike se utilizan otras dos proteasas como blanco de la drogas. Se utilizó el paquete MOPAC 2012 de química cuántica (QC) para generar el ranking de que tan «buenas» serían esas moléculas como ligandos de las proteínas de interés. El método es un desarrollo del grupo de Cavasotto y resulta novedoso respecto a la metodología más tradicional de scoring basado en mecánica clásica.
Estrategias de Cuarentena
Benjamín Marcolongo de FaMAF-UNC realizó simulaciones de estrategias de cuarentena basados en la circulación de personas. El nos cuenta de que se trata su trabajo:
Estoy usando Mendieta para hacer simulaciones de epidemias con modelos de agentes móviles que pueden transportar información -en este caso una enfermedad-. Es decir, simulamos personas -agentes móviles- que se mueven, interactúan -a través de choque- y que durante esa interacción pueden transmitir la enfermedad. Esto es, básicamente, el proceso concreto con el que las enfermedades se propagan.
Benjamín compara esta forma de atacar el problema con las llamadas Teorías de Campo Medio, donde el ejemplo que más conocemos es el modelo que utiliza R0 también conocido como Ritmo Reproductivo Básico, que puede depender de muchas cosas
El objetivo de la investigación es entender cuál es el rol de la movilidad o circulación de personas en la propagación de una enfermedad. También se busca explicar las fluctuaciones en el proceso epidémico a partir de estas circunstancias de movilidad. Marcolongo se pregunta:
¿Por qué en ciudades con características similares se pueden ver resultados tan diferentes? ¿Cómo se conecta la aleatoriedad del movimiento de los individuos en una sociedad con la probabilidad de que haya una epidemia? ¿Cómo varían los resultados respecto de distintas «estrategias» de movimientos? ¿Cuán sensible son las respuestas de los modelos a la implementación de cuarentenas -que se traducen en políticas de reducción de circulación-? ¿Cuál es el alcance y las limitaciones de este tipo de teorías para explicar fenómenos tan complejos como las epidemias? ¿Es posible predecir un pico y la duración de un pico? ¿Es posible asociar una teoría basada en distribuciones de probabilidades que sirva para hacer predicciones razonables? Son algunas de las preguntas que nos estamos haciendo y que en gran medida estamos empezando a entender.
El rol del CCAD-UNC es fundamental, ya que el proceso de simulación es computacionalmente intensivo.
La supercomputadora me permitió hacer miles de experimentos que son costosos. En cada simulación tengo que evolucionar en el tiempo una sociedad con todos sus individuos y con todas sus interacciones. Hacer tantos experimentos numéricos -que son necesarios para construir la estadística- con mi computadora de escritorio me hubiera llevado meses -tal vez años-. Con un nodo de Mendieta y paralelizando las simulaciones pude hacer -al día de la fecha- alrededor de 50 mil simulaciones de epidemias en distintas condiciones. Estoy convencido que sin este nodo mi investigación hubiera sido mucho más lenta y con resultados más difusos.
El software es un desarrollo propio en C++ que utilizar un red-black tree para optimizar la parte de las interacciones. Aunque el programa simula en un tiempo razonable 100.000 partículas, es decir el tamaño de una ciudad, se optó por un modelo más pequeño de 1000 agentes para poder hacer estadística, ya que los procesos son estocásticos. Marcolongo ensaya a manera de resumen.
Lo principal que vemos es que la movilidad es muy importante para entender si hay o no epidemias y las fluctuaciones son enormes. Es decir: si tirás una epidemia en la misma ciudad podés tener resultados muy diferentes.
Notas relacionadas
- «Las supercomputadoras de la UNC contra la Pandemia«, Marcela Palermo y Luis Ernesto Zegarra, Canal 10, SRT, 20200722.
- «Las supercomputadoras de la UNC ayudan a enfrentar la pandemia«, La Voz del Interior, 20200721.


